文書化と適格性評価
GMP & とGAMP Vのプロセス

適格性評価文書は、お客様の特定のニーズに適合させることができ、個別のアプローチが可能です。
必要なすべての文書の準備と管理は、クールバキュームの専門家がお客様に代わりすべて行います。
設計の文書化
- 適格性評価プロジェクト計画(QPP)
- 機能および設計仕様(FDS)
- ハードウェア設計仕様(HDS)
- ソフトウェア設計仕様(SDS)
- リスク評価(RA)
試験の文書化
- 設計時適格性評価(DQ)
- コンピュータシステムバリデーション(CSV)
- 工場受入試験(FAT)
- 現地受入試験(SAT)
- 設置および試運転(IAC)
- 設備据付時適格性評価(IQ)
- 運転時適格性評価(OQ)
- 発生した逸脱の記録
- 適格性評価変更管理の記録

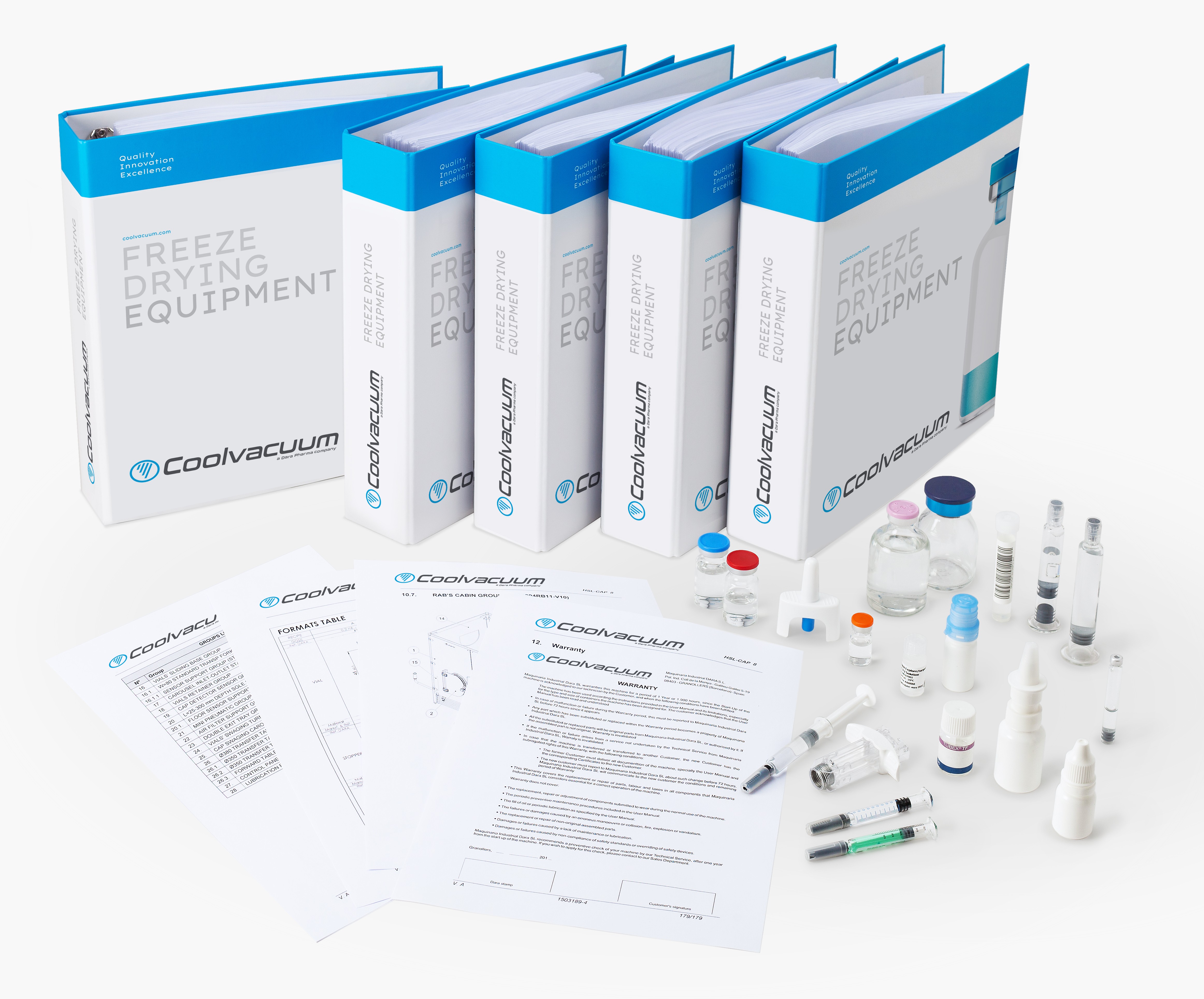
GMP装置の文書パッケージ。

品質管理。